In the lifecycle of equipment and system qualification, Design Qualification (DQ) is the initial yet most decisive stage. A minor design flaw undetected at this phase can lead to significant wasted costs for retrofitting and future compliance risks.
For investors and managers, DQ is more than just a compliance document; it is a strategic tool to control quality starting the "blueprint," ensuring the equipment is fit for its intended purpose and meets all regulatory standards.
1. What is DQ and Why Should Investors Care?
.jpg)
Design Qualification (DQ) is the documented process of verifying that the proposed design of equipment, systems, or facilities meets the User Requirement Specifications (URS) and regulatory standards (such as EU GMP, FDA).
Why is DQ critical?
- CAPEX Optimization: Identifying and fixing design flaws before fabrication or installation is significantly cheaper than rectifying them once the equipment is on-site.
- Ensuring Compliance: Confirming the design adheres to sanitary standards, cleanability, and cross-contamination prevention—vital elements in pharmaceutical manufacturing.
- Minimizing Operational Risk: Ensuring equipment meets productivity, safety, and performance expectations.
2. Strategic Objectives of the DQ Process
A robust DQ process must achieve the following:
- Verify the design precisely meets technical specifications and functional requirements.
- Identify and address potential design flaws affecting performance, safety, or product quality.
- Establish a foundational record for subsequent validation stages (IQ, OQ, PQ) and regulatory audits.
- Ensure the system is capable of producing safe, effective, and high-quality products.
3. Standard DQ Implementation Roadmap
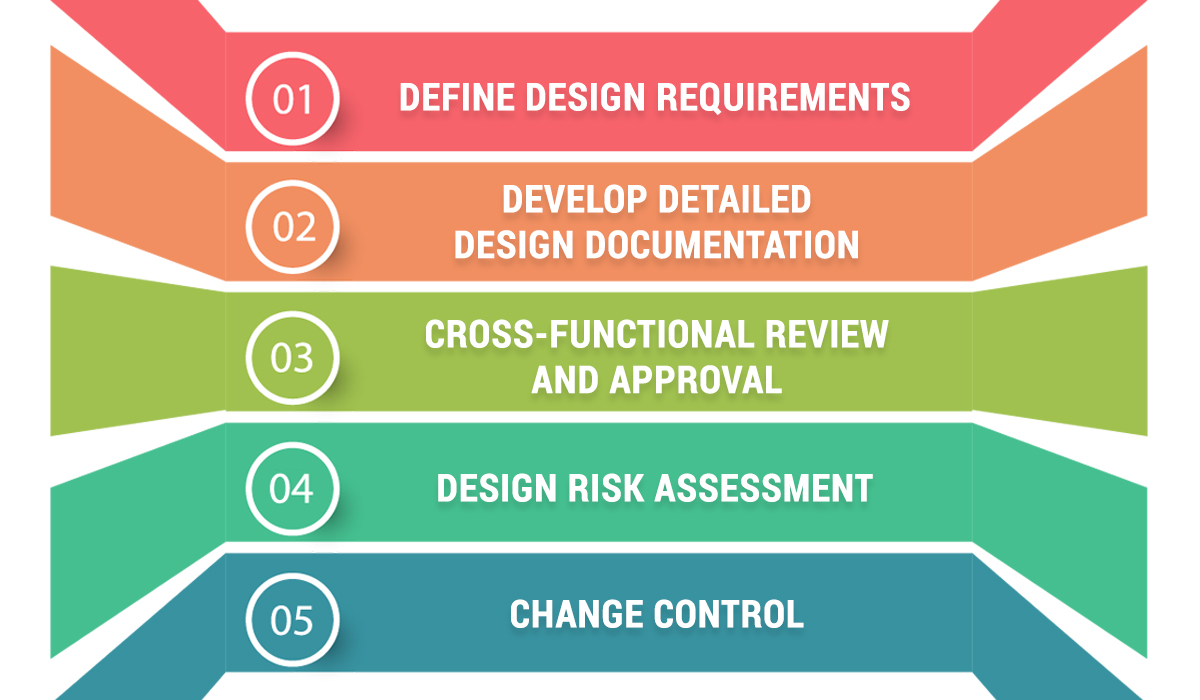
To make DQ an effective management tool, the following steps are required:
- Define Design Requirements: Establish performance criteria, GMP standards, and specific intended use.
- Develop Detailed Design Documentation: Including engineering drawings, material specifications, P&IDs, and other technical records.
- Cross-Functional Review and Approval: Design documents must be reviewed by a multi-disciplinary team, including Engineering, Production, and especially Quality Assurance (QA).
- Design Risk Assessment: Use tools like FMEA to identify potential design risks and implement mitigation measures (e.g., material changes or added safety features).
- Change Control: Any design modifications after DQ approval must be strictly managed to ensure consistency and compliance.
4. Management Perspective: DQ as Operational Insurance
DQ is not an isolated administrative procedure but an integral part of the broader verification and validation process. Investing seriously in the DQ stage allows business owners to:
- Shorten Time-to-Market: Avoid technical issues that cause delays during installation and commissioning.
- Build a Quality Culture: Demonstrate a commitment to compliance the very first stages of investment.
Conclusion
Design Qualification (DQ) is the first and most vital safeguard to ensuring that pharmaceutical systems and equipment are built correctly the start. A rigorous DQ process protects not only patient safety but also the corporate return on investment.

