Trong lộ trình đầu tư nhà máy dược phẩm, giai đoạn Thẩm định và Nghiệm thu (Commissioning & Qualification - C&Q) luôn là "điểm nghẽn" (bottleneck) tiêu tốn nhiều nguồn lực nhất. Đây là khoảng thời gian rủi ro cao khi dòng tiền đầu tư liên tục chi ra nhưng nhà máy vẫn chưa thể sản xuất thương mại để tạo doanh thu.
Thực tế tại nhiều dự án ở Việt Nam, tư duy "làm hồ sơ đối phó" hoặc thực hiện thẩm định theo quy trình cũ rườm rà đang khiến chủ đầu tư lãng phí hàng tháng trời cho việc kiểm tra lặp lại không cần thiết.
Bài viết này của GMPc Việt Nam sẽ phân tích phương pháp tiếp cận C&Q hiện đại: tập trung vào quản trị rủi ro để loại bỏ các thủ tục dư thừa, giúp dự án về đích đúng hạn (Time-to-Market) mà vẫn đảm bảo tính tuân thủ tuyệt đối.
1. Sự Chuyển Dịch Tư Duy: Từ Mô Hình V-Model Cổ Điển Sang C&Q Tích Hợp
Để hiểu được chiến lược mới, cần nhận diện hạn chế của cách làm cũ.
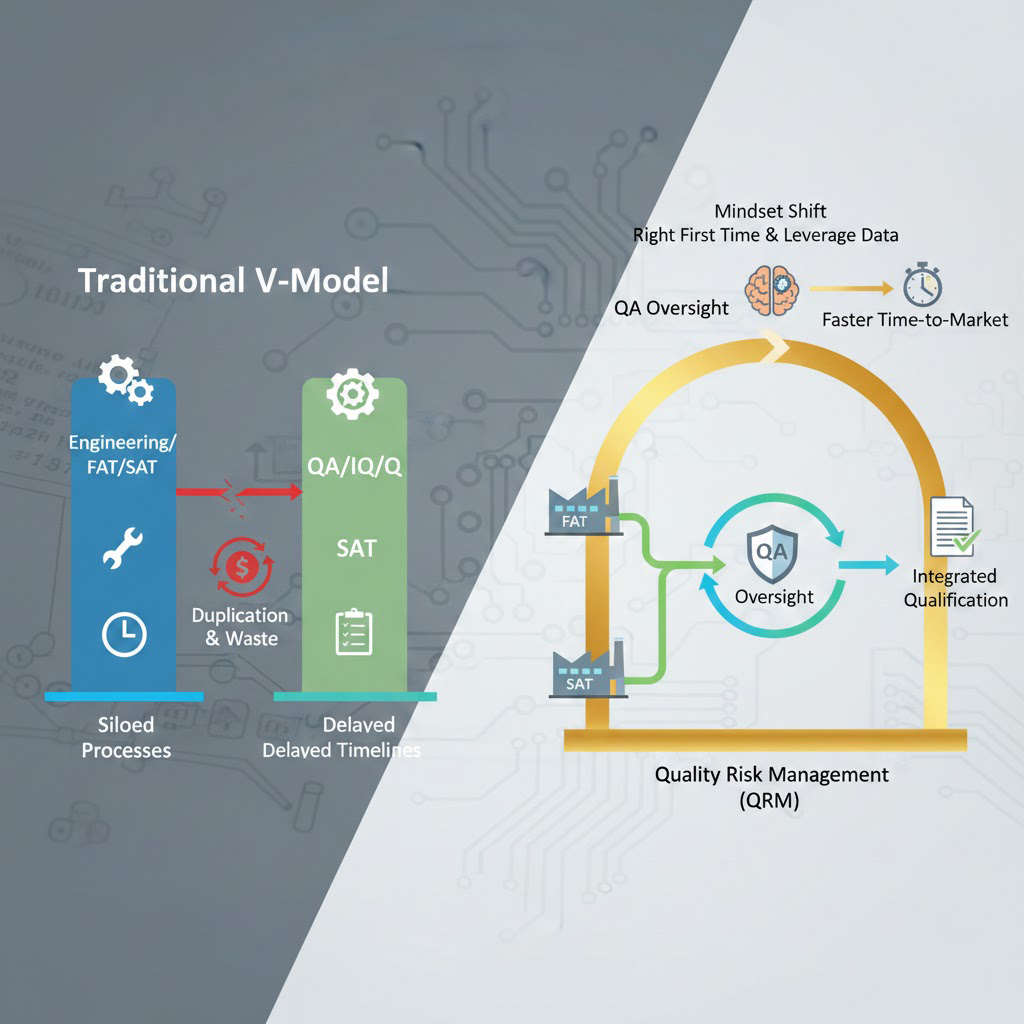
1.1. Cách tiếp cận truyền thống (Traditional V-Model)
Trong mô hình này, các giai đoạn Nghiệm thu kỹ thuật (Commissioning) và Thẩm định (Qualification) thường được tách biệt hoàn toàn.
- Hệ quả: Việc kiểm tra nghiệm thu thu máy móc (FAT/SAT) do bộ phận Kỹ thuật/Nhà thầu thực hiện. Sau đó, bộ phận QA lại thực hiện lại các bài test tương tự trong giai đoạn IQ/OQ để "lấy số liệu làm hồ sơ".
- Lãng phí: Sự lặp lại không cần thiết (Duplication of effort), kéo dài thời gian dự án và gia tăng chi phí nhân sự.
1.2. Cách tiếp cận hiện đại (Integrated C&Q / ASTM E2500)
Tiêu chuẩn EU GMP hiện đại khuyến khích áp dụng phương pháp Quản lý rủi ro chất lượng (QRM).
- Nguyên tắc: "Làm đúng ngay từ đầu và tận dụng dữ liệu".
- Chiến lược: Tích hợp các hoạt động Commissioning và Qualification. Dữ liệu thu được từ giai đoạn FAT (Nghiệm thu tại nhà máy chế tạo) và SAT (Nghiệm thu tại hiện trường) nếu được thực hiện đúng quy trình và có sự giám sát của QA, sẽ được sử dụng trực tiếp cho báo cáo Thẩm định mà không cần làm lại.
2. Quy Trình C&Q Tiêu Chuẩn Cho Nhà Máy EU GMP
Một lộ trình C&Q bài bản cần tuân thủ trình tự logic sau để đảm bảo tính tuân thủ và hiệu quả:
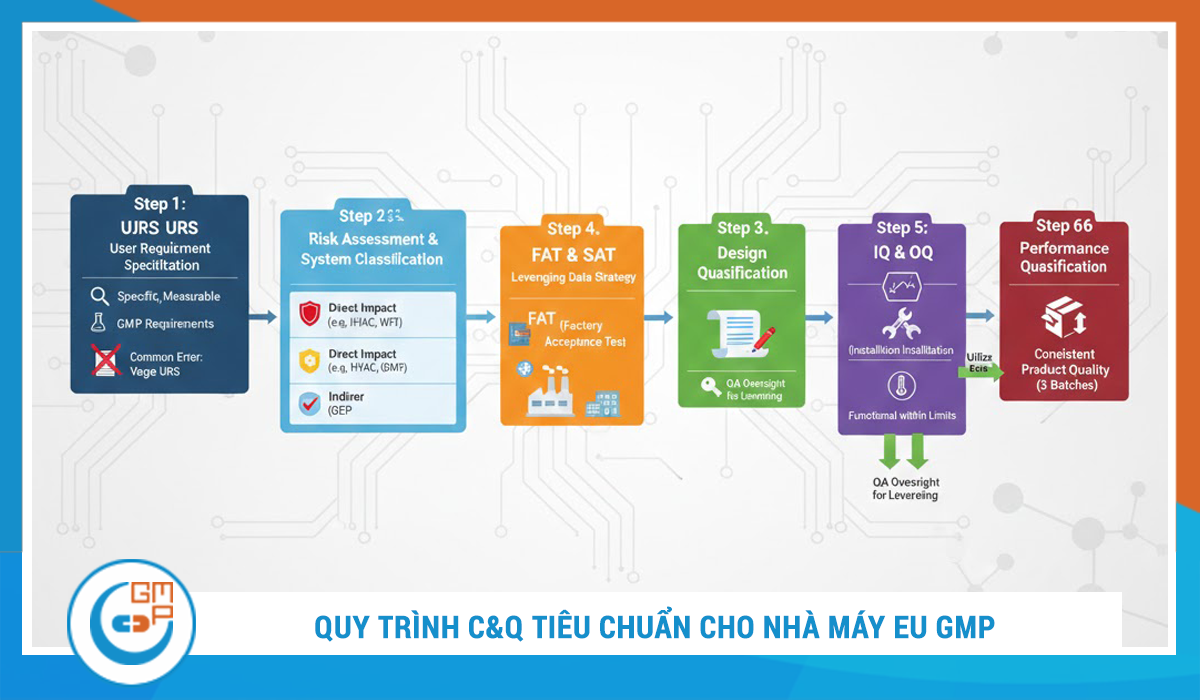
Bước 1: URS - Bản Tuyên Ngôn Của Dự Án (User Requirement Specification)
Đây là tài liệu quan trọng nhất, là "đề bài" mà Chủ đầu tư đặt ra cho nhà cung cấp.
- Yêu cầu: URS phải cụ thể, đo lường được và gắn liền với các yêu cầu GMP (Ví dụ: Nhiệt độ kho 15-25°C, độ đồng đều ±1°C).
- Lỗi thường gặp: URS sơ sài dẫn đến việc mua sai thiết bị hoặc thiết bị không đáp ứng được yêu cầu kiểm nghiệm sau này.
Bước 2: Đánh Giá Rủi Ro & Phân Loại Hệ Thống (System Classification & Risk Assessment)
Không phải thiết bị nào cũng cần thẩm định giống nhau. Cần phân loại hệ thống thành:
- Hệ thống tác động trực tiếp (Direct Impact): Ảnh hưởng trực tiếp đến chất lượng thuốc (Ví dụ: HVAC, Nước cất pha tiêm, Máy dập viên) $rightarrow$ Cần Thẩm định đầy đủ (Full Qualification).
- Hệ thống tác động gián tiếp (Indirect Impact): (Ví dụ: Chiller, Nồi hơi) $rightarrow$ Chỉ cần Nghiệm thu kỹ thuật (Good Engineering Practice - GEP).
- Không tác động (No Impact): (Ví dụ: Hệ thống thoát nước mưa).
Bước 3: DQ - Thẩm Định Thiết Kế (Design Qualification)
Rà soát lại bản thiết kế kỹ thuật và thiết kế chi tiết (Detailed Design) xem có đáp ứng URS và tiêu chuẩn GMP hay không trước khi đặt hàng chế tạo.
Bước 4: FAT & SAT - Tận Dụng Dữ Liệu (Leveraging Strategy)
- FAT (Factory Acceptance Test): Kiểm tra tại xưởng nhà sản xuất. Đây là cơ hội tốt nhất để phát hiện lỗi phần cứng/phần mềm.
- SAT (Site Acceptance Test): Kiểm tra sau khi lắp đặt tại nhà máy.
- Điểm mấu chốt: QA cần tham gia duyệt đề cương và giám sát quá trình FAT/SAT để biến các dữ liệu này thành bằng chứng thẩm định, giảm tải cho bước IQ/OQ.
Bước 5: IQ & OQ - Thẩm Định Lắp Đặt & Vận Hành
- IQ (Installation Qualification): Xác nhận thiết bị được lắp đặt đúng thiết kế, đúng tài liệu kỹ thuật.
- OQ (Operational Qualification): Xác nhận thiết bị vận hành ổn định trong các giới hạn thiết kế (bao gồm cả chạy thử tải cao nhất/thấp nhất, kiểm tra các chức năng cảnh báo/an toàn).
Bước 6: PQ - Thẩm Định Hiệu Năng (Performance Qualification)
Chứng minh hệ thống hoạt động ổn định và cho ra sản phẩm đạt chất lượng đồng nhất trong điều kiện sản xuất thực tế (thường thực hiện với 3 lô liên tiếp hoặc chạy liên tục trong một khoảng thời gian quy định).
3. Thẩm Định Vệ Sinh (Cleaning Validation) & Thẩm Định Quy Trình (Process Validation)
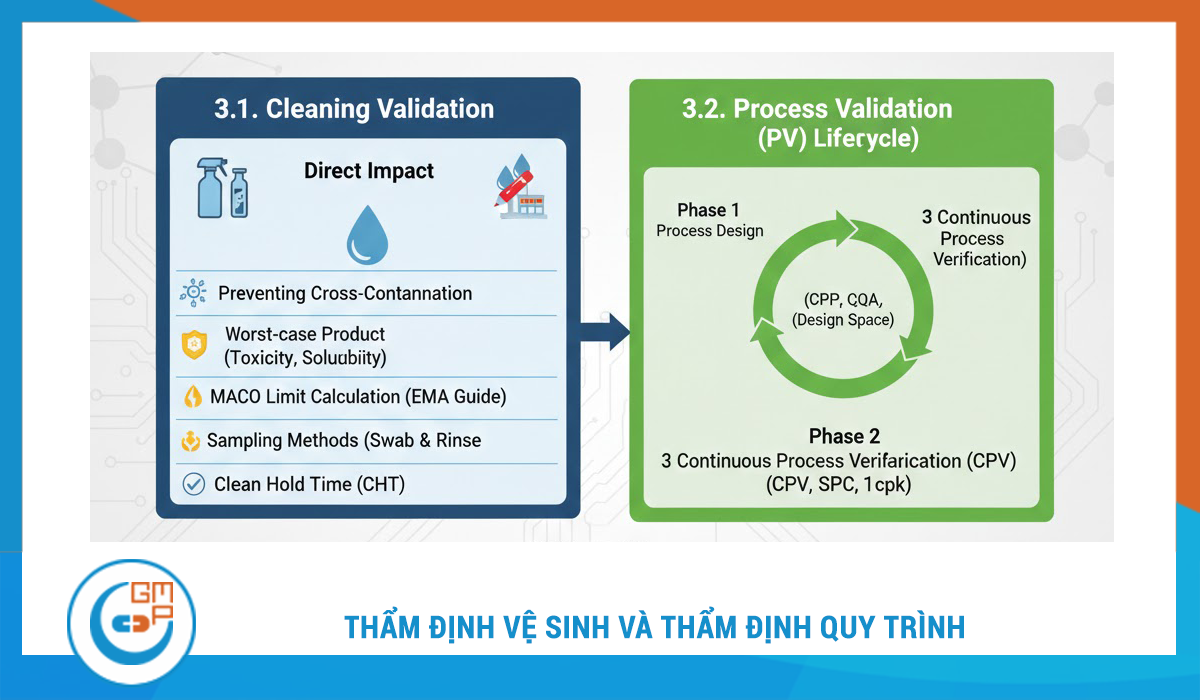
Sau khi hệ thống thiết bị (Hardware) đã sẵn sàng, nhà máy cần thực hiện thẩm định các yếu tố phi vật lý:
- Thẩm định vệ sinh: Chứng minh quy trình vệ sinh loại bỏ được dư lượng hoạt chất và chất tẩy rửa xuống dưới mức an toàn (MACO - Maximum Allowable Carryover). Đây là yêu cầu bắt buộc để kiểm soát Nhiễm chéo.
- Thẩm định quy trình: Theo hướng dẫn mới của EMA và FDA, thẩm định quy trình không chỉ là "3 lô thẩm định" mà là một vòng đời liên tục (Process Validation Lifecycle):
1. Thiết kế quy trình (Process Design).
2. Đánh giá quy trình (Process Qualification).
3. Kiểm chứng quy trình liên tục (CPV - Continued Process Verification).
4. Lợi Ích Chiến Lược Khi Áp Dụng Mô Hình Mới
Việc áp dụng mô hình C&Q tích hợp theo ISPE mang lại giá trị định lượng rõ rệt cho Chủ đầu tư:
1. Rút ngắn thời gian (Time-to-Market): Giảm 20-30% thời gian giai đoạn thực hiện dự án nhờ loại bỏ các công việc trùng lặp.
2. Tiết kiệm chi phí: Giảm chi phí nhân sự và chi phí vận hành thử nghiệm.
3. Hồ sơ khoa học: Hệ thống hồ sơ được xây dựng dựa trên logic rủi ro, dễ dàng giải trình với thanh tra viên EU/PIC/S thay vì một đống giấy tờ rập khuôn.
5. Kết Luận & Vai Trò Của Tư Vấn
C&Q không phải là đích đến, mà là phương tiện để chứng minh sự kiểm soát. Một chiến lược C&Q thông minh sẽ biến giai đoạn này từ "gánh nặng" thành "lợi thế cạnh tranh", giúp nhà máy sớm đi vào khai thác thương mại và thu hồi vốn.
Tại GMPc Việt Nam, chúng tôi không chỉ cung cấp dịch vụ soạn thảo tài liệu. Chúng tôi đóng vai trò là Quản trị dự án C&Q, hỗ trợ Chủ đầu tư:
- Xây dựng Kế hoạch tổng thể thẩm định (VMP).
- Đánh giá năng lực nhà cung cấp thiết bị.
- Giám sát và thực hiện trực tiếp các quy trình DQ/IQ/OQ/PQ.
- Đào tạo đội ngũ nhân sự nội bộ tiếp nhận và duy trì hệ thống tái thẩm định (Re-validation).
Chủ đề liên quan:
Hướng dẫn thiết kế và thẩm định hệ thống phòng sạch nhà máy dược phẩm (HVAC, Nước, Khí nén)
Toàn Vẹn Dữ Liệu (Data Integrity) Trong Nhà Máy EU GMP: Hướng Dẫn Thực Thi Toàn Diện & Chiến Lược Số Hóa
Nhân Sự Vận Hành Nhà Máy EU GMP: Thách Thức Và Giải Pháp Quản Trị Dành Cho Chủ Đầu Tư Tại Việt Nam
DỊCH VỤ TƯ VẤN & THỰC HIỆN THẨM ĐỊNH (C&Q) NHÀ MÁY EU GMP
Quý doanh nghiệp cần tư vấn chiến lược C&Q cho dự án nhà máy EU GMP vui lòng liên hệ tại đây.





