Việc đáp ứng nguyên tắc “Thực hành tốt sản xuất thuốc” (GMP) là yêu cầu bắt buộc đối với mọi cơ sở sản xuất thuốc và nguyên liệu làm thuốc nhằm đảm bảo chất lượng, an toàn và hiệu quả của sản phẩm dược. Thông tư 28/2025/TT-BYT vừa ban hành là căn cứ pháp lý quan trọng mới nhất, quy định rõ hồ sơ, trình tự, quy trình và phương pháp đánh giá mức độ tuân thủ GMP. Dưới đây là hướng dẫn chuyên sâu dành cho các doanh nghiệp và cá nhân liên quan trong lĩnh vực dược phẩm.
I. Hồ Sơ Làm Căn Cứ Để Đánh Giá Đáp Ứng GMP
1. Thành phần hồ sơ GMP căn cứ theo Luật Dược và Nghị định 163/2025/NĐ-CP

Cơ sở sản xuất nộp hồ sơ đề nghị cấp Giấy chứng nhận đủ điều kiện kinh doanh dược, đồng thời là cơ sở để đánh giá đáp ứng GMP, bao gồm:
- Tài liệu theo khoản 1 và điểm a khoản 2 Điều 20 của Nghị định 163/2025/NĐ-CP: bao gồm thông tin pháp lý, điều kiện cơ sở vật chất, nhân sự, trang thiết bị...
- Trường hợp sản xuất thuốc phải kiểm soát đặc biệt, bổ sung tài liệu tại điểm a khoản 1 Điều 37 Nghị định 163.
- Hồ sơ tổng thể kỹ thuật của cơ sở sản xuất (theo hướng dẫn tại Phụ lục VIII của Thông tư 28) hoặc hồ sơ cập nhật khi mở rộng phạm vi hoạt động.
2. Lưu ý quan trọng
- Nếu đồng thời xin cấp giấy chứng nhận GMP cùng với giấy đủ điều kiện kinh doanh dược, cần ghi rõ nội dung này trong Đơn đề nghị.
- Nếu cơ sở có bán/giao thuốc cho đơn vị phân phối hoặc cơ sở khám chữa bệnh, cần nộp thêm hồ sơ kỹ thuật và nhân sự theo nguyên tắc GDP (Thực hành tốt phân phối thuốc).
- Miễn trừ hồ sơ GDP nếu giao hàng ngay tại kho của cơ sở sản xuất.
II. Trình Tự Đánh Giá Việc Đáp Ứng GMP
1. Tiếp nhận hồ sơ
Cơ sở sản xuất nộp 01 bộ hồ sơ, kèm phí thẩm định đến cơ quan tiếp nhận của Bộ Y tế, tùy theo loại sản phẩm:
| Loại sản phẩm |
Cơ quan tiếp nhận |
|
Dược liệu, thuốc cổ truyền
|
Cục Quản lý Y, Dược cổ truyền
|
|
Nguyên liệu làm thuốc, thuốc hóa dược, dược liệu, vắc xin, sinh phẩm
|
Cục Quản lý Dược
|
|
Sản xuất phối hợp nhiều loại
|
Cục Quản lý Dược
|
2. Trình tự tiếp nhận và xử lý hồ sơ
- Bước 1: Nhận hồ sơ và cấp Phiếu tiếp nhận hồ sơ (Mẫu số 01).
- Bước 2: Trong 5 ngày làm việc, Cơ quan tiếp nhận thành lập Đoàn đánh giá và gửi quyết định cho cơ sở.
- Bước 3: Trong 7 ngày làm việc (hoặc 3 ngày nếu thuộc diện ưu tiên), Đoàn đánh giá thực hiện đánh giá thực tế tại cơ sở.
III. Quy Trình Đánh Giá Việc Đáp Ứng và Phân Loại Mức Độ Tuân Thủ GMP
1. Tài liệu sử dụng khi đánh giá
- Tiêu chuẩn WHO-GMP
- Tiêu chuẩn GMP do Bộ Y tế quy định tại Điều 3 Thông tư 28/2025/TT-BYT, tùy theo từng loại hình sản xuất.
2. Quy trình đánh giá tại cơ sở thực tế
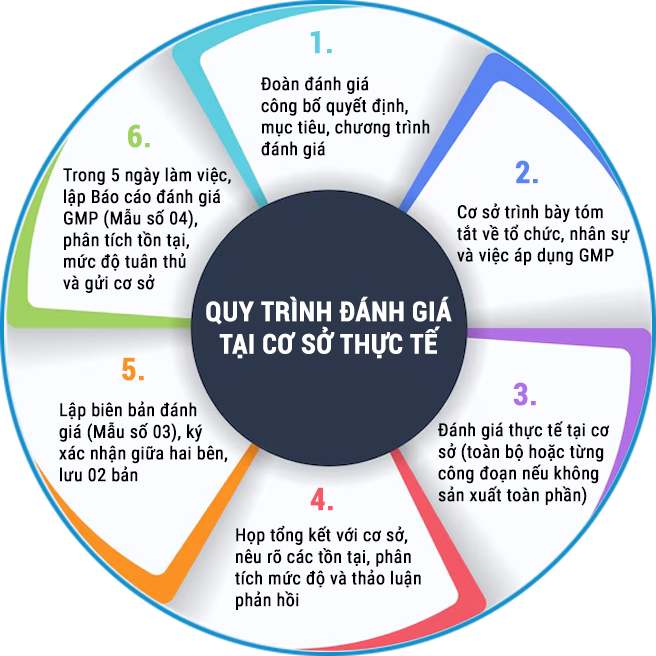
3. Phân loại mức độ tuân thủ GMP (theo Phụ lục IX)
- Mức 1: Tuân thủ đầy đủ GMP, không có tồn tại hoặc tồn tại nhỏ, không ảnh hưởng chất lượng
- Mức 2: Có tồn tại trung bình nhưng không ảnh hưởng lớn đến chất lượng, có khả năng khắc phục
- Mức 3: Tồn tại nghiêm trọng, ảnh hưởng đến chất lượng hoặc an toàn, cần khắc phục trước khi được cấp phép
- Mức 4: Không đáp ứng GMP, không đủ điều kiện sản xuất
Kết Luận
Thông tư 28/2025/TT-BYT quy định rõ ràng và chặt chẽ về hồ sơ, trình tự và quy trình đánh giá đáp ứng GMP, là căn cứ pháp lý cốt lõi giúp ngành dược nâng cao chuẩn mực sản xuất. Các doanh nghiệp dược phẩm cần chủ động cập nhật nội dung mới, chuẩn bị kỹ hồ sơ và phối hợp tốt với cơ quan quản lý để được đánh giá và phân loại ở mức độ cao nhất – qua đó tạo lợi thế cạnh tranh trong thị trường dược phẩm ngày càng khắt khe.
Quý khách hàng cần hỗ trợ cụ thể trong việc soạn hồ sơ tổng thể, chuẩn bị trước đánh giá GMP hoặc đào tạo nội bộ áp dụng tiêu chuẩn GMP mới nhất, đội ngũ chuyên gia của GMPc Việt Nam sẵn sàng đồng hành. Liên hệ ngay để được tư vấn chi tiết và đúng chuẩn pháp lý!
So sánh thông tư 28/2025/TT-BYT với thông tư 35/2018/TT-BYT về Thực hành tốt sản xuất thuốc, nguyên liệu làm thuốc
Thông tư 28/2025/TT-BYT quy định về Thực hành tốt sản xuất thuốc (GMP), nguyên liệu làm thuốc





